
Xover is an online bioinformatic tool for automatic analysis and plotting of DNA/protein crossover patterns. It can analyze complex DNA/protein mosaic patterns generated by various DNA chimeragenesis technologies such as DNA shuffling fast and accurately without human intervention, and present data in a graph that is not only rich in information but also easy to understand. Xover is part of the online computational suite ReX. See the other tool of ReX: REcut.
Please cite: Huang W., Johnston W.A., Boden M. and Gillam E.M.J. (2016) ReX: A suite of computational tools for the design, visualization, and analysis of chimeric protein libraries. Biotechniques, 60(2):91-94. (PMID: 26842355)
Paste FASTA sequences of parents into the top textbox.
Paste FASTA sequences of chimeras into the bottom textbox.
Both DNA and protein are supported. Pre-alignment is not required. You may submit up to 200 KB data, which allows about 200 sequences of 1 kb DNAs or 600 sequences of 300 aa proteins.
By default, the crossover graph will be drawn in pre-defined colors. Alternatively, you can try the Random Colors option.
Statistics such as the means and standard deviations of crossovers and point mutations, library quality indicators, and a compressed crossover graph will be displayed in a few seconds. A high resolution uncompressed crossover graph can be downloaded in BMP format for publication quality using the link shown at the bottom of the result page. The link to the full dataset used for generating the graph can also be found at the bottom of the page for further analysis if necessary.
A cytochrome P450 3A subfamily DNA shuffling library created using 3A4, 3A5, 3A7 and 3A9 as parents is used in the demonstration. After sequence submissions of parent DNAs and chimera DNAs, you can see a crossover graph and corresponding library statistics in a few seconds. An example result page is provided below:
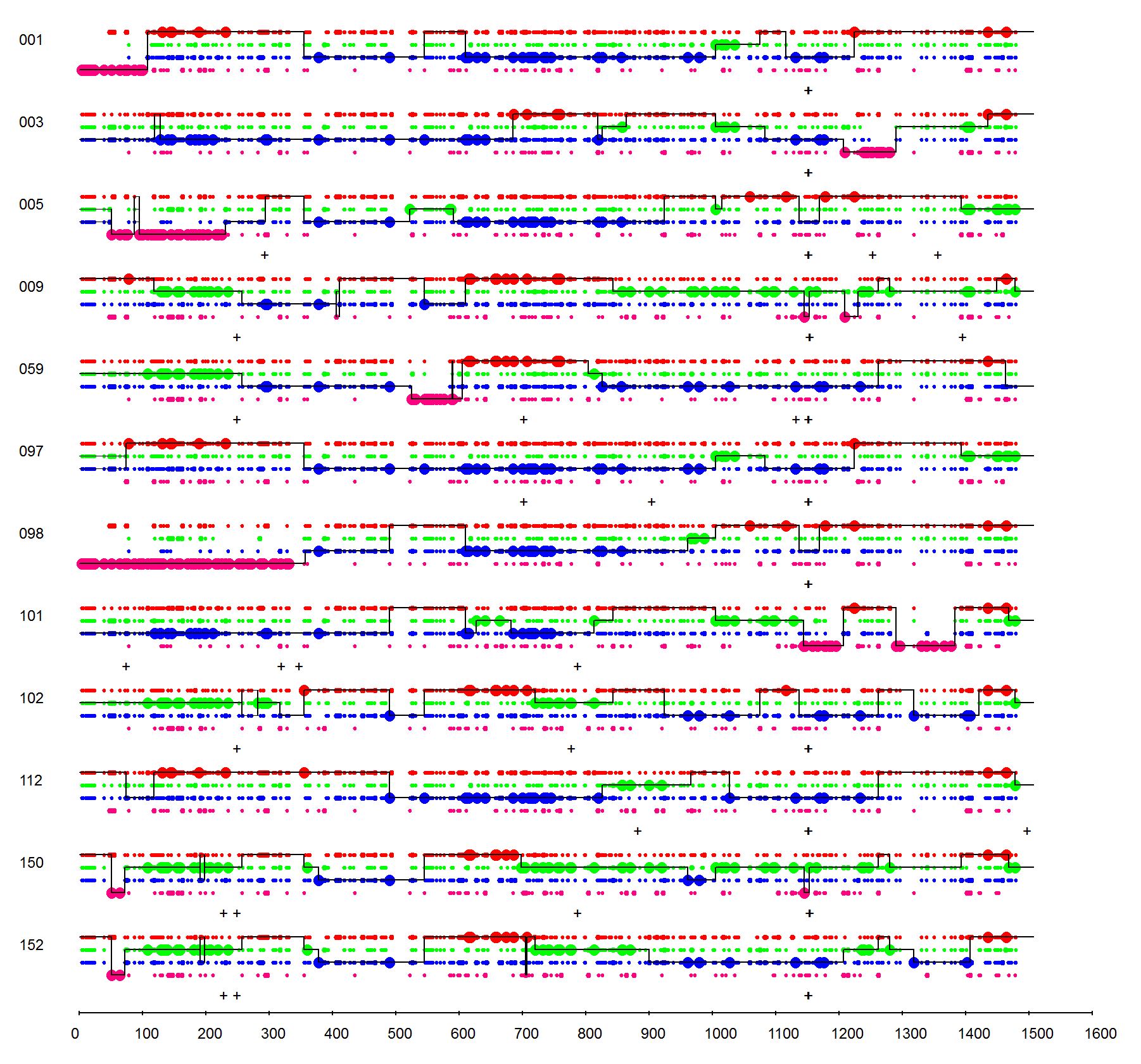

On the top of the result page, essential statistics of the library are provided, such as the means and standard deviations of crossovers and point mutations. A figure legend is displayed after the statistics, which explains the drawing and symbols used in the graph. Briefly, library parents are depicted in different colors in order of entry of the parents. Large dots represent 100% parental match (i.e. the position in question matches only one parent) and small dots represent more than one parental match (i.e. the position matches more than one parent) at each position. The solid line for each chimera represents the library parents identified within the sequence between crossovers. A set of horizontal parallel lines between crossovers indicates multiple parents match at an equal probability (e.g. 5' ends of mutants 003 and 005 around 100 bp). A mutation is recorded as a plus sign when a position of a chimera does not match the corresponding position in any parental sequence. A vertical spike indicates a fast single position switch between parents (e.g. the spike downwards in the middle of mutant 152 around 700 bp). A horizontal black bar indicates a fragment insertion/deletion.
To evaluate relationship between mutants and parents, the real mutations introduced by DNA shuffling into each mutant is measured by Levenshtein distance. Levenshtein distance measures sequence difference between mutants and parents, i.e. minimum point mutations required to convert a parent into a mutant. A parent showing the shortest distance is the closest parent to the mutant. This distance is called Effective mutation, i.e. the minimum point mutations required to back-mutate a mutant to its closest parent. To evaluate the diversity of the library, Effective mutation is used to measure the actual diversity introduced into each mutant.
For fast display of large graphs over the internet, a compressed graph is shown on the result page, which might have subtle image quality loss sometimes. For publication purpose, you can download the full resolution lossless graph in BMP format using the link at the bottom of the result page. Meanwhile, the full dataset that has been analyzed by the program can also be downloaded. It contains all the information and analysis data that is used to plot the crossover graph, including sequence alignments, per-position parent calling, point mutation detection, detailed crossover information. The data is stored in CSV format, which can be readily open in any spreadsheet programs such as Microsoft Excel.
You can plot not only the DNA sequences of mutants but also the protein sequences of your library.
Pre-aligned sequences are not necessary. Xover will do all the work for you fully automatically.
If you don't like the default colors scheme, try re-plotting using the Random Colors option.
If you submit more than 100 sequences in a batch, it might take more than 1 minute to show the result. Please be patient and don't refresh your browser or resubmit the same batch before the result page is displayed.
Xover supports very large crossover graphs, however you might want to split your library sequences in a few batches and submit 10~20 chimeras each time to allow the generated graph to fit the page size of Office software easily.
Extremely narrow (spike-like) crossovers on the crossover graph are caused by point mutations, which accidentally are an existing nucleotide or amino acid that can also be found in another parent at the same position, sometimes. Another possible source of these spikes is extremely frequent template switching during fragment extension by PCR.
Point mutations observed throughout the library at the same position of sequences are usually inherited from the parent DNA templates. They can be confirmed by sequencing the parent DNAs used to construct the library.
Incomplete ORFs, raw contig assemblies and mutants showing low levels of sequence insertions/deletions/truncations can be used for a quick evaluation of library usually. However, high quality full length sequences are strongly recommended for the formal characterization and final publication. Low quality sequences and mutants with significant sequence disruptions could distort the final aggregated results significantly and should be analyzed individually.
If you have any question or need any assistance, please don't hesitate to contact:
Your suggestions are always welcome.